
Adrenogenitales Syndrom mit 21-Hydroxylase-Defekt (AGS)
Unter dem kongenitalen (= angeborenen) adrenogenitalen Syndrom (AGS) werden mehrere von beiden Eltern vererbte Defekte der Cortisolbiosynthese der Nebenniere zusammengefasst.
Ursache
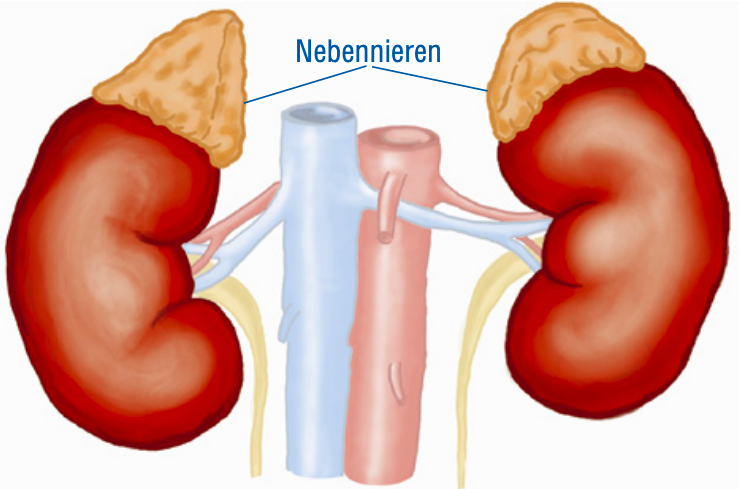
Die Nebenniere ist eine kleine Drüse, die der Niere wie eine Zipfelmütze aufsitzt und verschiedene lebenswichtige Hormone produziert. Die wichtigsten Hormone sind das „Stresshormon” Cortisol, das im Energiehaushalt sowie im Zucker- und Fettstoffwechsel mitwirkt, das Aldosteron, das an der Regulierung des Salz- und Wasserhaushaltes und damit des Blutdrucks beteiligt ist, und die Androgene (Androstendion, Testosteron), verantwortlich für Haarwuchs und Muskelzuwachs.
Die unterschiedlichen Formen des AGS werden durch genetische Störungen der an der Biosynthese beteiligten Enzyme verursacht. In über 95 % aller Fälle ist das Enzym 21-Hydroxylase betroffen. In diesem Fall kann Progesteron nicht mehr zu 11-Desoxycorticosteron und 17-Hydroxy-Progesteron nicht mehr zu 11-Desoxycortisol verstoffwechselt werden; die Produktionskette ist an dieser Stelle unterbrochen. Ein Kontrollsystem erkennt, dass ein Fehler vorliegt (das heißt, dass zu wenige Hormone nach dem Enzymdefekt produziert werden) und meldet dies der Hirnanhangsdrüse (Hypophyse). Diese versucht die Nebenniere anzuregen, um die Produktion der Hormone aufrechtzuerhalten. Die Stimulation bewirkt aber keine effektive Hormonproduktion, sondern nur, dass sich die Nebennieren vergrößern und die Hormonvorstufen vor dem Produktionsstop durch den Enzymdefekt anstauen. Daher wird die Erkrankung auch angeborene Nebennierenhyperplasie genannt.

Da die Nebenniere aber nur die Hormone vor dem Stopp bilden kann, sammeln sich immer mehr Vorstufen an. Das Hormon vor dem Stopp ist beim 21-Hydroxylasemangel das 17-Hydroxy-Progesteron (17-OH-P). Dieses Hormon kann im Blut und Speichel gemessen werden. Ein großer Teil dieses Hormons und anderer Vorstufen wird ungenutzt ausgeschieden und und ist im Urin messbar. Ein anderer Teil wird durch Enzyme in einen anderen Stoffwechselweg eingeschleust, wodurch vermehrt Androgene (= männliche Hormone) produziert werden. Man findet also beim AGS mit 21-Hydroxylase-Defekt klinische Symptome der Nebennieren-Unterfunktion und Symptome der vermehrten Produktion männlicher Hormone.
Der Einfluss von Cortisol, Aldosteron und Androgene

Formen des AGS
Das AGS mit 21-Hydroxylase-Defekt tritt in zwei Formen, der klassischen und der nicht-klassischen Form auf.
Symptome
Fast alle betroffenen Mädchen mit klassischem AGS haben bei Geburt aufgrund der in der Gebärmutter stattfindenden Vermännlichung ein vermännlichtes äußeres Genitale. Das innere Genitale ist immer weiblich. Beim AGS kommt es bei Mädchen daher auch zu einer Störung der Geschlechtsdifferenzierung, die als 46,XX-DSD oder Variante bezeichnet wird. „DSD” steht für Disorder of Sexual Development und „46, XX” gibt den normalen weiblichen Chromosomensatz an. Heutzutage wird statt DSD auch öfter der Begriff „Variante der Geschlechtsentwicklung“ verwendet. Der Schweregrad der Vermännlichung wird nach Prader in 5 Stadien eingeteilt. Die Veränderungen können von einer einfachen Vergrößerung der Klitoris (Klitorishypertrophie (Prader 1)) bis hin zur kompletten Fusion der Labioskrotalfalten mit einer penisartig vergrößerten Klitoris und Ausweitung der Harnröhre auf die Eichel (Prader 5) reichen. Die weiblichen Neugeborenen können daher bei Geburt als Knaben verkannt werden.

Diagnose
Das AGS wird laborchemisch, molekulargenetisch und klinisch durch die körperliche Untersuchung diagnostiziert.
Wachstum und Pubertät

Eine entscheidende Rolle spielt die Qualität der Therapie, denn sowohl eine zu niedrige als auch eine zu hohe Glukokortikoid-Substitutionsdosis haben negative Auswirkungen auf das Längenwachstum. Aktuelle Studien zeigen, dass die Erwachsenengröße der Patienten meist unter der Zielgröße liegt. Beginn und Verlauf der Pubertät sind nahezu normal; bei guter Einstellung ist das Alter beim ersten Auftreten der Menstruation normal.
AGS und Schwangerschaft

Bei einer guten medikamentösen Einstellung kann fast jeder an AGS erkrankte Patient Kinder bekommen. Bevor man sich dazu entschließt, muss aber unbedingt eine ausführliche genetische Beratung erfolgen. Man muss vor allem unterscheiden, ob ein klassisches AGS oder ein nicht-klassisches AGS vorliegt. Der Partner/die Partnerin muss auf das Vorliegen eines heterozygoten AGS untersucht werden, das heißt, ob er Überträger der Erkrankung ist. Dazu sollte er sich bei einem Spezialisten (Genetiker) zusammen mit dem betroffenen Partner vorstellen. Es wird dann in der Regel eine molekulargenetische Untersuchung durchgeführt. Ist Ihr Partner genetisch gesund, so ist das gemeinsame Kind nur Überträger des klassischen bzw. nicht- klassischen AGS. Wenn Sie selbst ein klassisches AGS haben und Ihr Partner hat ein heterozygotes AGS mit einer Mutation, die eine geringe Restaktivität der 21-Hydroxylase bedingt, so wird das Kind mit 50-prozentiger Wahrscheinlichkeit an einem klassischen AGS erkranken. Wenn Sie aber ein nicht-klassisches AGS haben und Ihr Partner ist z. B. heterozygoter Genträger für das nicht- klassische AGS mit einer Mutation, die eine höhere Restaktivität der 21-Hydroxylase bedingt, so wird das Kind mit 50-prozentiger Wahrscheinlichkeit an einem nicht-klassischen AGS erkranken.
Die Therapie des AGS wird in der Schwangerschaft selbstverständlich fortgeführt. Besteht für den Fötus kein AGS-Risiko, dann sollte während der Schwangerschaft kein Dexamethason gegeben und die Therapie rechtzeitig auf ein nicht-plazentagängiges Glukokortikoid umgestellt werden. Während der Schwangerschaft ist eine engmaschige Überwachung erforderlich, um die Substitutionsdosis an die jeweilige Situation (eventuell Dosiserhöhung in den letzten drei Monaten, unter der Geburt) anzupassen.
Behandlung und Therapie

Durch den Mangel an Hormonen besteht beim klassischen AGS die Notwendigkeit einer Substitutionstherapie (= Zuführung der fehlenden natürlichen Hormone von außen durch Tabletten). Diese Therapie muss ein Leben lang beibehalten werden. Im Kindesalter wird in der Regel mit dem natürlichen körpereigenen Hormon Hydrocortison (= Cortisol) behandelt. Die Tagesdosis (ca. 10– 15 mg/m2 Körperoberfläche) wird üblicherweise in 3 Dosen verabreicht. Da die Cortisolproduktion bei Gesunden frühmorgens am höchsten ist, wird der größte Teil der Tagesdosis von Hydrocortison (50 %) morgens gegeben. Bei Erwachsenen wird die Hormonersatztherapie mit Hydrocortison meist beibehalten. Manchmal wird jedoch nach Ende des Wachstums auf eine Therapie mit einem Glukokortikoid mit einer längeren Verweildauer im Blut (z. B. Prednisolon, Dexamethason) übergegangen. Mittlerweile gibt es auch ein Hydrocortison mit verzögerter Wirkstoff-Freisetzung, das extra für Menschen mit AGS entwickelt wurde, das nur noch zweimal am Tag (morgens und vor dem Schlafen) eingenommen werden muss. Da Cortisol ein „Stresshormon” ist, wird es in Belastungssituationen des Körpers in bis zu 5-facher Menge ausgeschüttet. Dieser natürlichen Reaktion des Körpers muss die Medikamentengabe angepasst werden. Können aus irgendeinem Grund keine Tabletten geschluckt werden, müssen die Glukokortikoide im Notfall gespritzt (Vene oder Muskel) oder auch einmalig per Zäpfchen (rektal) verabreicht werden. Jeder Patient muss einen Notfallausweis erhalten.
Es gibt einen Europäischen Cortisolmangel-Notfallausweis im Scheckkartenformat, der beim betreu- enden Endokrinologen ausgegeben werden sollte, alternativ ist dieser auch über die Selbsthilfegruppen erhältlich.
Übergang vom Jugendalter zum Erwachsenenalter

Manche Jugendliche mit AGS haben eventuell noch nicht gelernt, Eigenverantwortung zu übernehmen, weil die Eltern noch für die Medikamenteneinnahme sorgen, die Therapie überwachen und Arzttermine vereinbaren. Viele der Patienten haben nie eine schwere Salzverlustkrise erlebt, die zu einer stationären Behandlung geführt hat, sodass sie die Notwendigkeit der regelmäßigen Tabletteneinnahme hinterfragen. Probleme mit der Compliance, also der Therapietreue, sind in der Pubertät bei chronisch Kranken häufig. Zusätzlich scheinen die hormonellen Veränderungen in der Pubertät die Wirkung von Hydrocortison zu beeinträchtigen. Die Folge ist eine zum Teil dramatische Verschlechterung der Therapieeinstellung.
Haben die Jugendlichen und die Eltern das AGS – als eine lebenslange Erkrankung – verstanden und angenommen, dann sollten beim Übergang von der Pädiatrie in die Erwachsenenmedizin eigentlich keine Probleme auftreten. Es müsste klar sein, dass die Betreuung durch einen internistischen Endokrinologen fortzusetzen ist.
")